
Заболевания с аутосомно-рецессивным типом наследования возникают только в случае, если ребенку передаются сразу оба гомологичных (одинаковых) мутантных гена : один от матери, другой от отца. Такой ребёнок окажется гомозиготным носителем мутантного гена, но его родители, являясь гетерозиготными носителями, вполне здоровы.
Следует помнить, что два гетерозиготных носителя одного и того же мутантного гена чаще встречаются среди родственников. Поэтому при родственном браке всегда имеется высокая вероятность родить больного ребёнка с наследственным заболеванием. Заболевания с таким типом наследования называют аутосомно-рецессивным. Они характеризуются наличием мутаций в одном и том же гене на одной и той же паре гомологичных хромосом (аутосом). К настоящему времени описано более 3000 наследственных заболеваний с аутосомно-рецессивным типом наследования.
Кратко рассмотрим некоторые наиболее часто встречающиеся актосомно-рецессивные заболевания, диагностика которых актуальна уже на саммых ранних стадиях развития, то есть ещё до рождения ребёнка.
К ним относится:
- муковисцидоз
- фенилкетонурия
- спинальная амиотрофия Верднига- Гоффмана
- адрено-генитальный синдром
- гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова)
Эти заболевания, повторяем, наблюдаются только в том случае, если оба родителя больного являются гетерозиготными носителями одного и того же мутантного гена. С вероятностью 25% они рискуют иметь больного ребёнка, причём вероятность его рождения не зависит от очередности беременности и от пола. Мальчики и девочки при заболевании с аутосомно-рецессивным типом наследования болеют одинаково часто, то есть никакого предпочтения полу при этом типе наследования не отмечается.
К сожалению, из множества моногенных аутосомно-рецессивных патологий успешное симптоматическое лечение разработано всего для нескольких десятков. Одним из них является гепатолентикулярная дегенерация (ГЛД). В отечественной литературе это заболевание также называют гепатоцеребральная дистрофия, или болезнь Вильсона, болезнь Вильсона-Коновалова. При ГЛД наблюдается генетически обусловленное нарушение усвоения (утилизации) солей меди вследствие мутации гена, кодирующего синтез белка, ответственного за обмен меди в организме. Этот белок был идентифицирован только несколько лет назад с помощью методов «обратной» генетики. Сначала ген был картирован на хромосоме 13 (13q14-q21), а затем и выделен. Полученные данные о молекулярной структуре гена и идентификация в нем различных мутаций открывают широкие возможности для молекулярной диагностики ГЛД в любом возрасте ( в том числе и в период внутриутробного развития предполагаемого больного), а так же позволяют проводить точное выявление гетерозиготных носителей патологического гена в семьях высокого риска по данному заболеванию.
На рисунке ниже представлена родословная больной с аутосомно-рецессивным типом наследования, в частности с ГЛД.
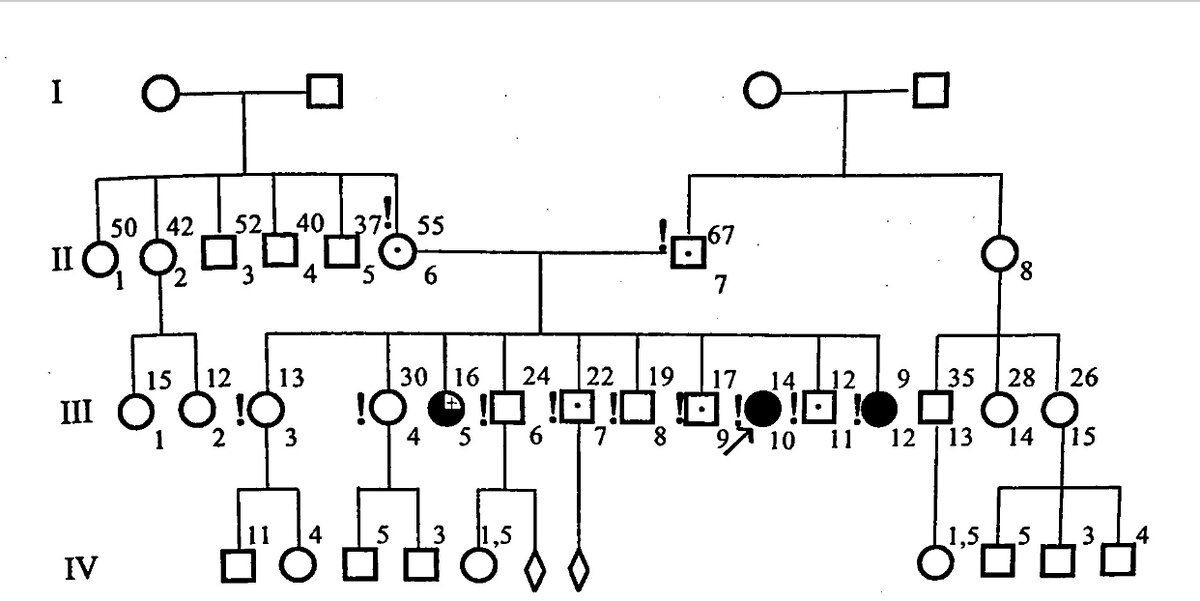 Родословная А-й (III-10), больной ГЛД
Родословная А-й (III-10), больной ГЛД Символы, применяющиеся при составлении.
Символы, применяющиеся при составлении.Из 10 детей в семье трое больны ГЛД, причём независимо от очерёдности рождения.
У больных ГЛД соли меди не утилизируются, а накапливаются клетками организма. Патологические накопления меди в печени, подкорковых структурах головного мозга могут быть причиной хронического гепатита (так называемого вильсоновского хронического гепатита) и подкорковой экстрапирамидной недостаточности. Последняя проявляется специфическим дрожанием конечностей и головы, возникающим при любых физических нагрузках и движениях. Поражение печени проявляется в среднем в 12,0 +\- 8,3 лет, головного мозга - в 23,0+\- 6,1 лет. Форма при которой наблюдаются неврологические признаки ГЛД, встречается значительно чаще, чем печёночная.
На рисунке ниже фотография больной ГЛД с типичным вильсоновским дрожанием рук, так называемым «flapping tremor” (“взмах крыльев птицы”). Дрожания нет при полном покое.
 Фото (В. Богатырева)
Фото (В. Богатырева)В 40-60-е годы прошлого столетия в СССР считалось, что причиной ГЛД является осложнение после перенесённого больным острого вирусного гепатита (ранее это инфекционное заболевание называли болезнью Боткина). Известно, что в указанные годы генетические исследования в стране не проводились, точнее- они были повсеместно запрещены из-за преступной антинаучной деятельности Т. Лысенко, возглавившего «крестовый поход» против вейсманизма-морганизма-менделизма. До 60-х годов последним сообщением в отечественной печати о наследственном происхождении ГЛД и аутосомно-рецессивном типе наследования, в частности, была глава об этом страдании в книге С.Н. Давиденкова «Наследственные болезни», изданной в 1925 году в Харькове.
В начале 60-х годов вышел в свет научно-фантастический приключенческий роман «Лезвие бритвы» писателя -фантаста Ивана Ефремова. Приводим выдержку из романа, касающуюся ГЛД.
«И вдруг красивый и здоровый юноша, способный музыкант, шахматист, заболел. Вялость, быстрая утомляемость и боли в правом боку быстро сменились расстройством походки, плохой координацией движений рук, сильными головными болями. Долго искали причину, юноша перекочевал уже в третью больницу и ему становилось только хуже. Сифилис исключен был сразу, опухоль мозга тоже. … Нашёлся умный врач и велел сделать анализ мочи на металлические соединения и обнаружил … медь. Следовательно, Вильсонова болезнь. … Болезнь Вильсона - от чего она бывает? Только наследственность тому виной».
Далее автор рассуждает о возможных механизмах наследственных болезней. Мы сейчас хорошо знаем, что при аутосомно-рецессивном типе наследования практически здоровые родители являются гетерозиготными носителями совершенно одинакового патологического рецессивного гена и их ребёнок с вероятностью 25% может иметь патологию, вызванную мутацией этого гена. Впервые в отечественной литературе (к сожалению, не специальной) появилось сообщение о том, что у героя И. Ефремова наблюдается в крови малая концентрация церулоплазмина - медьсодержащего белка. Справедливости ради, отмети, что церулоплазмин был открыт в 1948 году, а в 1952 году, американские учёные С. Шейнберг и Дж. Гитлин показали, что одним из характерных признаков ГЛД является резкое снижение уровня в сыворотке крови этого белка. Сделав вполне правильное заключение, что «недостаточность энзима (в данном случае- церулоплазмина) вызывает потерю меди, а эта потеря ведёт к поражению определённых участков мозга», И. Ефремов, следуя избранной для своего повествования истории, решил «перевернуть ситуацию». Он представил патогенез (механизм) болезни своего героя, как «поражение , или психическое подавление, тех участков мозга, которые ведут к изменению биохимии организма, потере этого самого энзима ( белка) и выделению меди». В данном случае такой механизм возникновения ГЛД не соответствует действительности. Может быть, писатель решил избрать психогенный механизм происхождения ГЛД для своего больного, так как в этом случае прогноз его лечения и жизни вполне благоприятный. И. Ефремов не знал, что ещё в 1956 году английский врач Уолш предложил при ГЛД применять связывающий и выводящий из организма ионы меди препарат пеницилламин. Эффективность этого чудодейственного при ГЛД лекарства отражена на рисунках ниже ⤵️
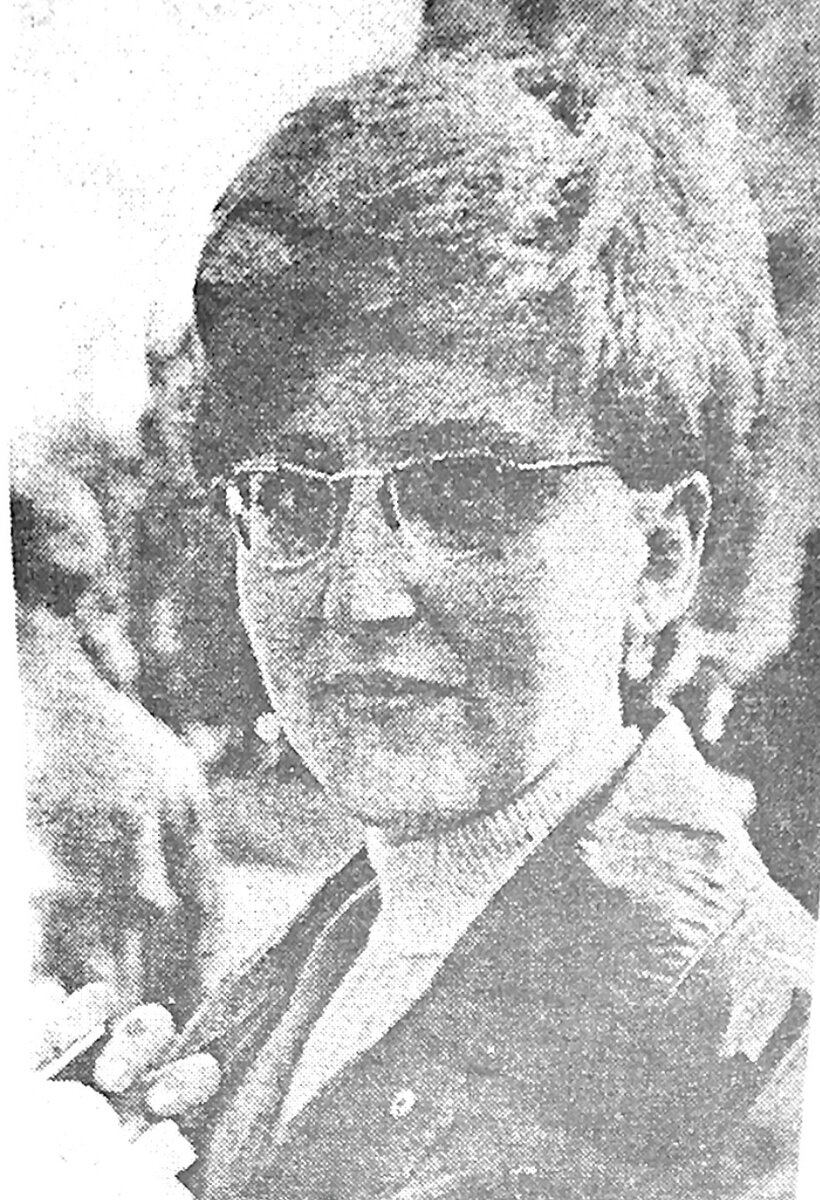 Больная Т., 31 год, ГЛД дрожательно-ригидной формы, за два года до начала неврологических проявлений заболевания.
Больная Т., 31 год, ГЛД дрожательно-ригидной формы, за два года до начала неврологических проявлений заболевания.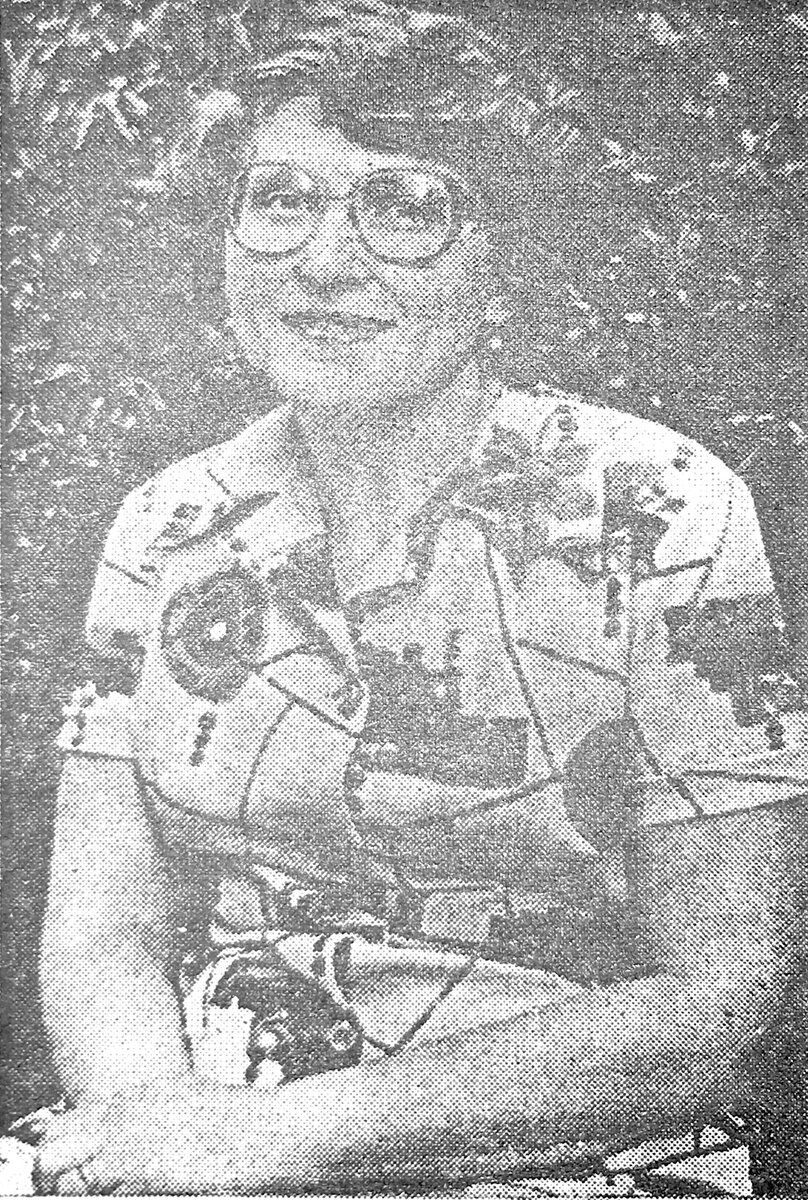 Та же больная через полтора года после начала лечения, включая применение пеницилламина.
Та же больная через полтора года после начала лечения, включая применение пеницилламина.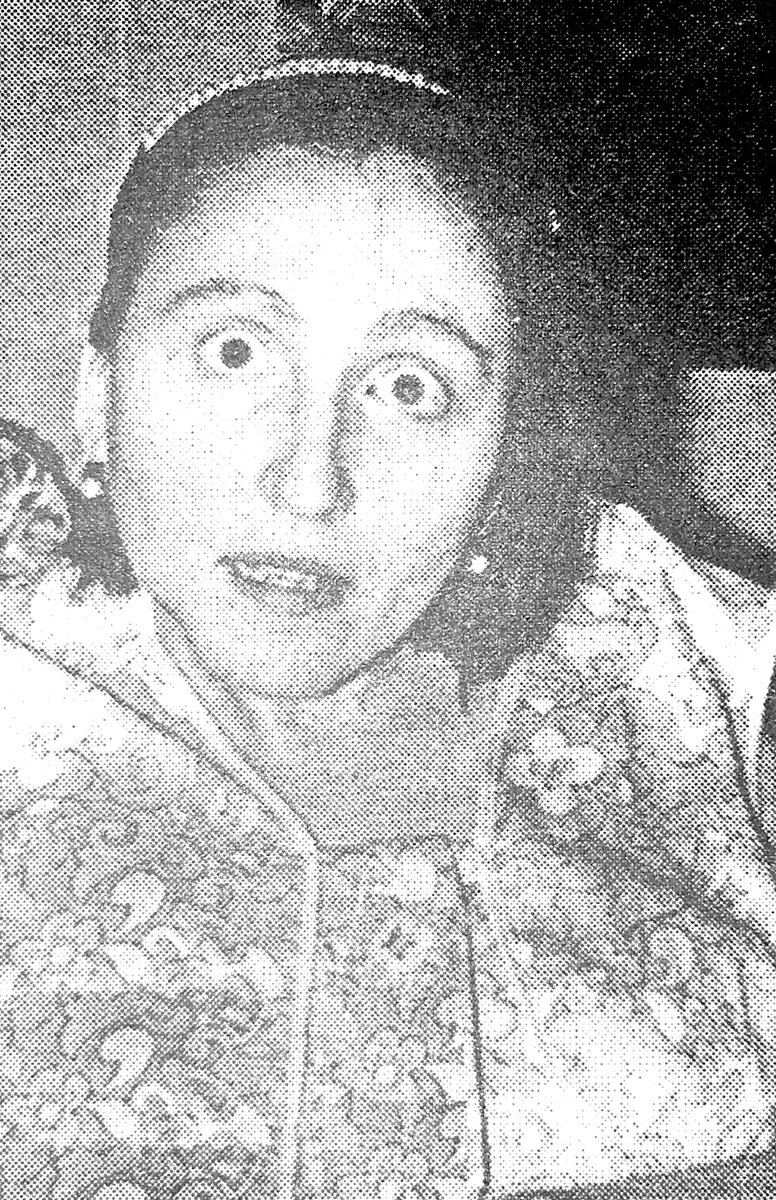 Та же больная за неделю до поступления под наблюдение докторов.
Та же больная за неделю до поступления под наблюдение докторов. Изменения почерка у больной Л., 27 лет, ГЛД дрожательной формы, на фоне лечения, включая применение пеницилламина : 1) почерк до лечения; 2) через 6 месяцев после начала лечения; 3) через 8 месяцев; 4) через полтора года; 5) через 3 года
Изменения почерка у больной Л., 27 лет, ГЛД дрожательной формы, на фоне лечения, включая применение пеницилламина : 1) почерк до лечения; 2) через 6 месяцев после начала лечения; 3) через 8 месяцев; 4) через полтора года; 5) через 3 годаТридцатый год больная Л., ( ее почерк в динамике на фоне лечения отражен на последнем рисунке) поддерживает оптимально стабильное состояние своего здоровья. Она аккуратнейшим образом до настоящего времени выполняет назначения врача. Серьезное заболевание является, безусловно, страданием для больного, лечение же- тяжёлым трудом. Наша больная умеет хорошо делать последнее. Применяя пеницилламин в доклиническом периоде болезни, можно предотвратить развитие тяжелой клинической симптоматики. В клинической стадии заболевания пеницилламин практически полностью избавляет от дрожания конечностей 85% больных, при условии обязательного выполнения назначений врача.
Отметим, что Уолш на основании изучения статуй Тутанхамона (египетский фараон в 1351-1342 до н.э. из 18 династии) в различные периоды его жизни высказал предположение, что фараон страдал или болезнью Клейнфелтера (писалось в ранних статьях про неё) или ГЛД. Если последнее верно, можно представить, сколько уже тысячелетий существуют мутации этого гена! В настоящее время частоту ГЛД оценивают как примерно три больных (гомозиготных носителей мутантного гена) на 100.000 здоровых, частота гетерозиготного носительства 1:200.
Мы так подробно рассматриваем ГЛД не только потому, что из немногих наследственных патологий эта поддаётся результативному лечению, но и потому, что диагноз заболевания можно установить у плода уже при сроке беременности 9-10 недель.
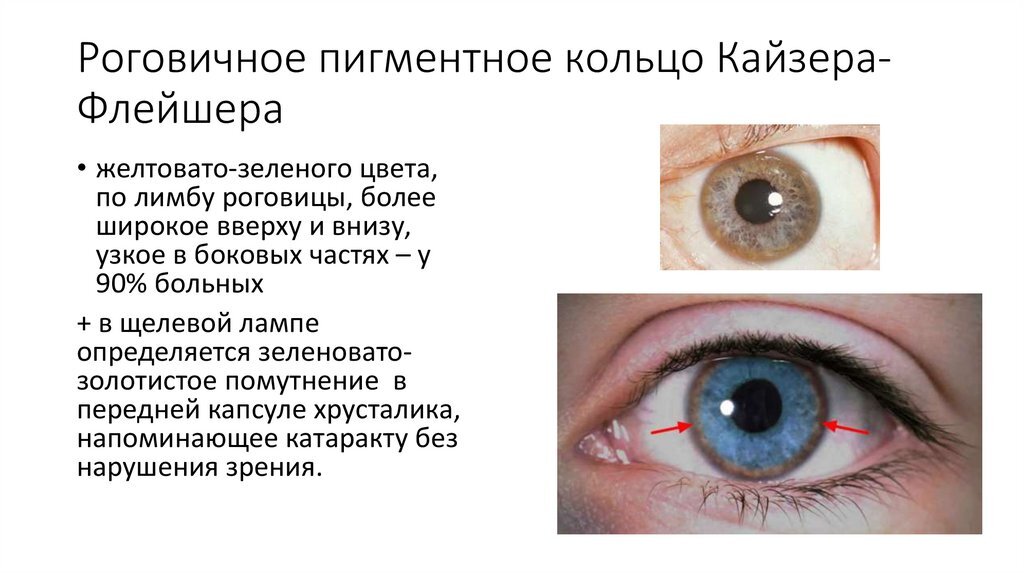
При наличии в семье одного больного ребёнка с ГЛД, как и в случае любого другого аутосомно-рецессивного заболевания, необходимо исключать и у больных в возрасте 10-30 лет с клинической картиной острого и хронического вирусного гепатита, но при отрицательных вирусологических тестах. В частности, исследовать уровень церулоплазмина в сыворотке крови, который при этом заболевании резко снижен, и проводить осмотр роговиц глаз через щелевую лампу с целью обнаружения специфических при ГЛД медных роговичных колец Кайзера-Флейшера. Отметим, что у детей кольца могут быть ещё не сформированы или выглядеть в виде дуг по периферии радужной оболочки.
Итак, повторяем.
Аутосомно-рецессивный тип наследования.
Родители, гетерозиготные носители рецессивного мутантного гена, практически здоровы. С риском 25% у них может родиться больной ребёнок.
Особо отмечаем:
в том случае, если в семье имеется двое и более детей с одинаковым по клинической картине тяжелым заболеванием, то таких больных необходимо исследовать на наличие патологии с аутосомно-рецессивным типом наследования, а родственников-тестировать на носительство патологического гена. При подозрении на наличие в семье больного с наследственной болезнью сам больной и его родственники должны быть непременно проконсультированы врачом-генетиком.
Спасибо за внимание! Про муковисцидоз, фенилкетонурию и др. Мы напишем в следующей статье.
#медицина в россии #медицинская энциклопедия #аутосомно-рецессивный тип наследования #мутант #родители #генетические мутации #генетика #ГЛД #родственные отношения #вирусный гепатит
диагностика
Больше интересных статей здесь: Медицина.
Источник статьи: Аутосомно-рецессивный тип наследования (глд).