
Заболевания, передающиеся по аутосомно-рецессивному типу, проявляются у ребёнка только при условии, что он унаследовал две копии мутантного гена — по одной от каждого родителя. Такой ребёнок становится гомозиготным по патологическому гену и заболевает. При этом его родители, будучи гетерозиготными носителями (имеющими одну нормальную и одну мутантную копию гена), обычно остаются клинически здоровыми.
Важно понимать, что вероятность встречи двух носителей одного и того же рецессивного гена значительно выше среди кровных родственников. Поэтому в родственных браках резко возрастает риск рождения ребёнка с наследственной патологией. На сегодняшний день описано свыше 3000 заболеваний, наследуемых по аутосомно-рецессивному механизму, при котором мутация локализована в одном и том же гене на паре гомологичных (неполовых) хромосом.
Ключевые особенности и распространённые заболевания
Рассмотрим некоторые наиболее часто встречающиеся аутосомно-рецессивные болезни, диагностика которых крайне важна на самых ранних этапах, в том числе пренатально.
К ним относятся:
- Муковисцидоз
- Фенилкетонурия
- Спинальная амиотрофия Верднига-Гоффмана
- Адреногенитальный синдром
- Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова)
Эти состояния развиваются только если оба родителя являются носителями идентичного мутантного гена. В такой паре риск рождения больного ребёнка составляет 25% при каждой беременности, независимо от пола будущего ребёнка или очерёдности родов. Мальчики и девочки болеют с одинаковой частотой, что характерно для аутосомного типа наследования.
Гепатолентикулярная дегенерация (ГЛД) как излечимый пример
К сожалению, эффективное симптоматическое лечение разработано лишь для немногих из множества моногенных аутосомно-рецессивных патологий. Одним из таких исключений является гепатолентикулярная дегенерация (ГЛД), также известная как болезнь Вильсона-Коновалова. В основе заболевания лежит генетически обусловленное нарушение метаболизма меди из-за мутации в гене, ответственном за синтез медь-транспортирующего белка. Этот ген был картирован на 13-й хромосоме (13q14-q21), а его последующее выделение и изучение мутаций открыло возможности для точной молекулярной диагностики ГЛД в любом возрасте, включая пренатальный период, а также для выявления носителей в семьях риска.
Клиническая картина и патогенез ГЛД
У больных ГЛД нарушена утилизация меди, что приводит к её токсическому накоплению в тканях, прежде всего в печени и базальных ганглиях головного мозга. Это вызывает развитие хронического гепатита («вильсоновский гепатит») и неврологических расстройств в виде экстрапирамидной недостаточности. Неврологические симптомы, включая характерный «хлопающий» тремор (flapping tremor), обычно проявляются позже печёночных — в среднем в 23 года против 12 лет. Неврологическая форма заболевания встречается чаще.
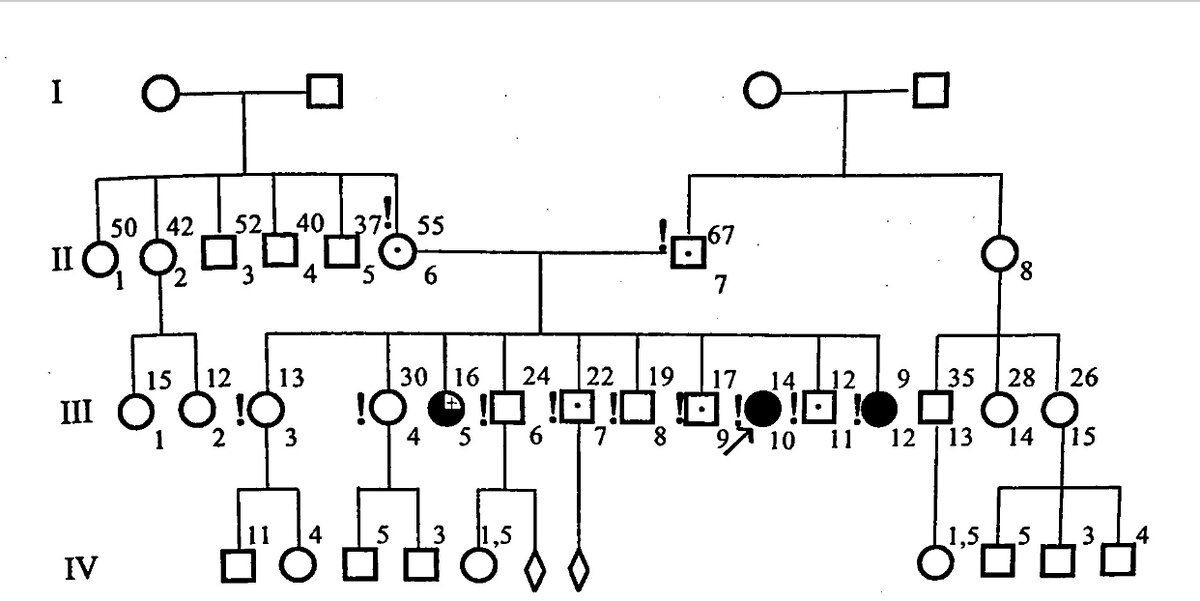
Родословная А-й (III-10), больной ГЛД. Из 10 детей в семье трое оказались больны, что наглядно демонстрирует случайный характер наследования при каждом зачатии.
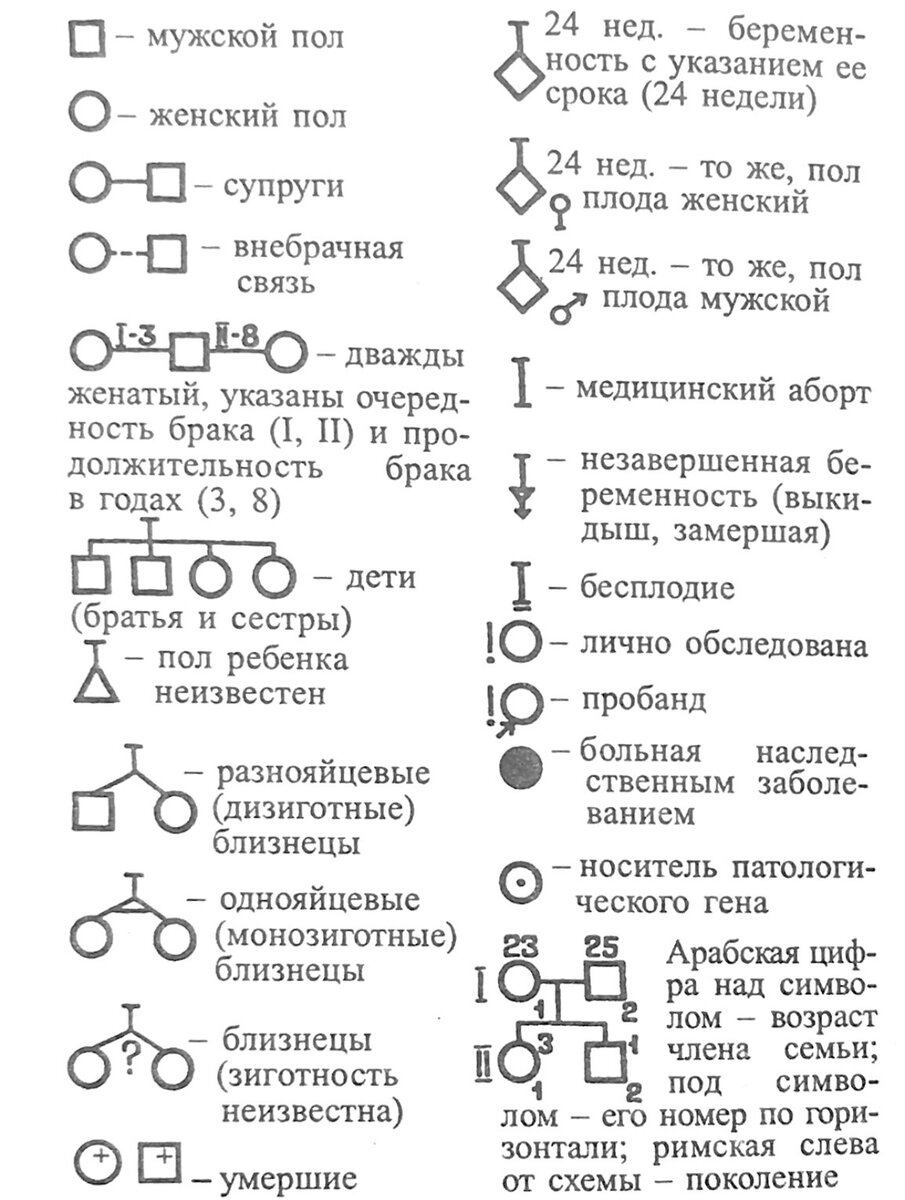
Символы, применяющиеся при составлении родословных.
История изучения и современная терапия
Интересна история изучения ГЛД в СССР, где генетические исследования долгое время были под запретом. В массовой культуре болезнь была описана Иваном Ефремовым в романе «Лезвие бритвы». Прорывом в лечении стало применение хелатора меди — пеницилламина, предложенного английским врачом Уолшем в 1956 году. Этот препарат способен связывать и выводить избыток меди из организма.
На примере пациентки Л., которая уже 30 лет поддерживает стабильное состояние благодаря дисциплинированному лечению, видна высокая эффективность терапии. Пеницилламин не только купирует симптомы у большинства больных, но и при назначении в доклинической стадии может предотвратить развитие болезни. Частота ГЛД оценивается как 3 случая на 100 000 человек, а носительство мутантного гена — примерно 1:200.
Диагностика и важность генетического консультирования
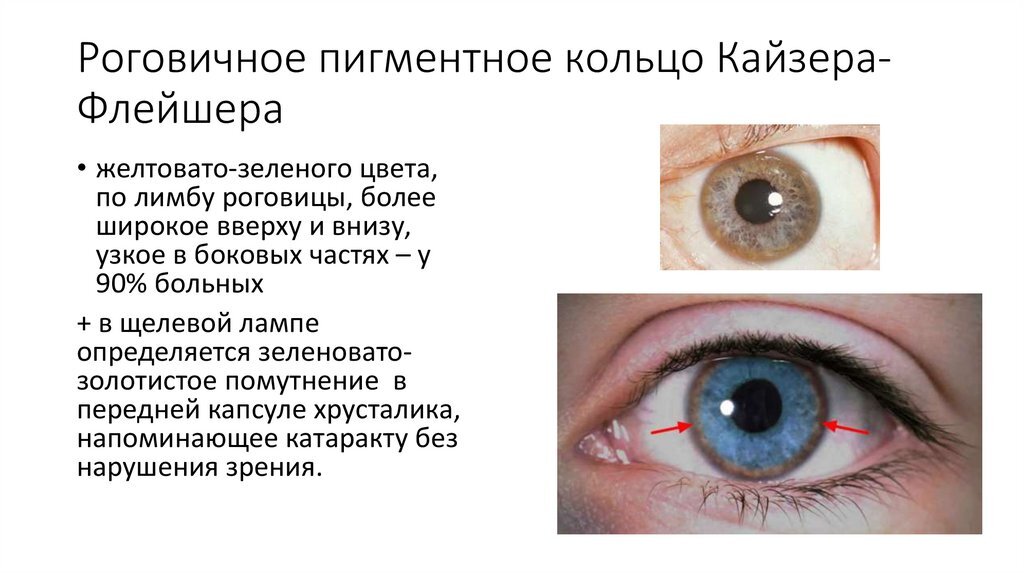
Диагностика ГЛД включает исследование уровня церулоплазмина в крови (он резко снижен) и осмотр роговицы глаза на наличие колец Кайзера-Флейшера. При наличии в семье ребёнка с ГЛД или любого другого аутосомно-рецессивного заболевания крайне важно обследовать родственников на носительство и проводить медико-генетическое консультирование.
Итоги: основные принципы аутосомно-рецессивного наследования
Родители — здоровые носители рецессивной мутации. Риск рождения больного ребёнка у такой пары — 25%. Если в семье есть двое и более детей со сходным тяжёлым заболеванием, необходимо исключать аутосомно-рецессивную патологию, тестировать родственников на носительство и обязательно направлять семью на консультацию к врачу-генетику.
В следующих статьях:
Мы подробнее расскажем о муковисцидозе, фенилкетонурии и других заболеваниях из этого списка.
#медицина в россии #медицинская энциклопедия #аутосомно-рецессивный тип наследования #мутант #родители #генетические мутации #генетика #ГЛД #родственные отношения #вирусный гепатит #диагностика
Больше интересных статей здесь: Медицина.
Источник статьи: Аутосомно-рецессивный тип наследования (глд).


